Related reading:
- Musings on Mechanism, Rob Phillips, https://www.ncbi.nlm.nih.gov/pubmed/28963318
- Excellent blog post on "Theoretical Amnesia" http://osc.centerforopenscience.org/2013/11/20/theoretical-amnesia/)

Mechanism. The word fills many of us with dread: “Not enough mechanism.” “Not particularly mechanistic.” "What's the mechanism?" So then what exactly do we mean by mechanism? I don’t think it’s an idle question—rather, I think it gets down to the very essence of what we think science means. And I think there are some practical consequences on everything from how we report results to the questions we may choose to study (and consequently to how we evaluate science). So I’ll try and organize this post around a few concrete proposals.
To start: I think the definition I’ve settled on for mechanism is “a model for how something works”.
I think it’s interesting to think about how the term mechanism has evolved in our field from something that really was mechanism once upon a time into something that is really not mechanism. In the old days, mechanism meant figuring out e.g. what an enzyme did and how it worked, perhaps in conjunction with other enzymes. Things like DNA polymerase and ATP synthase. The power of the hard mechanistic knowledge of this era is hard to overstate.
What can we learn about the power of mechanism/models from this example?
As the author of this post argues, models/theories are “inference tickets” that allow you to make hard predictions in completely new situations without testing them. We are used to thinking of models as being written in math and making quantitative predictions, but this need not be the case. Here, the predictions of how these enzymes function has led to, amongst other things, our entire molecular biology toolkit: add this enzyme, it will phosphorylate your DNA, add this other enzyme, it will ligate that to another piece of DNA. That these enzymes perform certain functions is a “mechanism” that we used to predict what would happen if we put these molecules in a test tube together, and that largely bore out, with huge practical implications.
Mechanisms necessarily come with a layer of abstraction. Perhaps we are more used to talking about these in models, where we have a name for them: “assumptions”. Essentially, there is a point at which we say, who knows, we’re just going to say that this is the way it is, and then build our model from there. In this case, it’s that the enzyme does what we say it will. We still have quite a limited ability to take an unknown sequence of amino acids and predict what it will do, and certainly very limited ability to take a desired function and just write out the sequence to accomplish said function. We just say, okay, assume these molecules do XYZ, and then our model is that they are important for e.g. transcription, or reverse transcription, or DNA replication, or whatever.
Fast forward to today, when a lot of us are studying biological regulation, and we have a very different notion of what constitutes “mechanism”. Now, it’s like oh, I see a correlation between X and Y, the reviewer asks for “mechanism”, so you knock down X and see less Y, and that’s “mechanism”. Not to completely discount this—I mean, we’ve learned a fair amount by doing these sorts of experiments, but I think it’s a pretty clear that this is not sufficient to say that we know how it works. Rather, this is a devolution to empiricism, which is something I think we need to fix in our field.
Perhaps the most salient question is what it does it mean to know “how it works?”. I posit that mechanism is an inference that connects one bit of empiricism to another. Let’s illustrate in the case of something where we do know the mechanism/model: a lever.
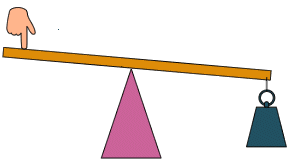
“How it works” in this context means that we need a layer of abstraction, and have some degree of inference given that layer of abstraction. Here, the question may be “how hard do I have to push to lift the weight?”. Do we need to know that the matter is composed of quarks to make this prediction, or how hard the lever itself is? No. Do we need to know how the string works? No. We just assume the weight pulls down on the string and whatever it’s made of is irrelevant because we know these to be empirically the case. We are going to assume that the only things that matter are the locations of the weight, the fulcrum, and my finger, as well as the weight of the, uhh, weight and how hard I push. This is the layer of abstraction the model is based on. The model we use is that of force balance, and we can use that to predict exactly how hard to push given these distances and weights.
How would a modern data scientist approach this problem? Probably take like 10,000 levers and discover Archimedes Law of the Lever by making a lot of plots in R. Who knows, maybe this is basically how Archimedes figured it out in the first place. It is perhaps often possible to figure out a relationship empirically, and even make some predictions. But that’s not what we (or at least I) consider a mechanism. I think there has to be something beyond pure empiricism, often linking very disparate scales or processes, sometimes in ways that are simply impossible to investigate empirically. In this case, we can use the concepts of force to figure out how things might work with, say, multiple weights, or systems of weights on levers, or even things that don’t look like levers at all. Wow!
Okay, so back to regulatory biology. I think one issue that we suffer from is that what we call mechanism has moved away from true “how it works” models and settled into what is really empiricism, sort of without us noticing it. Consider, for instance, development. People will say, oh, this transcription factor controls intestinal development. Why do they say that? Well, knock it out and there’s no intestine. Put it somewhere else and now you get extra intestine. Okay, but that’s not how it works. It’s empirical. How can you spot empiricism? A good sign is excessive obsession with statistics: effect sizes and p-values are often a good sign that you didn’t really figure out how it works. Another sign is that we aren’t really able to apply what we learned outside of the original context. If I gave you a DNA typewriter and said, okay, make an intestine, you would have no idea how to do it, right? We can make more intestine in the original context, but the domain of applicability is pretty limited.
Personally, I think that these difficulties arise partially because of our tools, but mostly because I think we are still focused on the wrong layers of abstraction. Probably the most common current layers of abstraction are those of genes/molecules, cells, and organisms. Our most powerful models/mechanisms to date are the ones where we could draw straight lines connecting these up. Like, mutate this gene, make these cells look funny, now this person has this disease. However, I think these straight lines are more the exception than the norm. Mostly, I think these mappings are highly convoluted in interwoven systems, making it very hard to make predictions based on empiricism alone (future blog post coming on Omnigenic Model to discuss this further).
Which leads me to a proposal: let’s start thinking about other layers of abstraction. I think that the successes of the genes/molecules -> cells paradigm has led to a certain ossification of thought centered around thinking of genes and molecules and cells as being the right layers of abstraction. But maybe genes and cells are not such fundamental units as we think they are. In the context of multicellular organisms, perhaps cells themselves are passive players, and rather it is communities of cells that are the fundamental unit. Organoids could be a good example of this, dunno. Also, it is becoming clear that genetics has some pretty serious limits in terms of determining mechanism in the sense I’ve defined. Is there some other layer involving perhaps groups of genes? Sorry, not a particularly inspired idea, but whatever, something like that maybe. Part of thinking this way also means that we have to reconsider how we evaluate science. As Rob pointed out, we have gotten so used to equating “mechanism” to “molecules and their effects on cells” that we have become both closed minded to other potential types of mechanism while also deceiving ourselves into allowing empiricism to pose as mechanism under the guise of statistics. We just have to be open to new abstractions and not hold everyone to the "What's the molecule?" standard.
Of course, underlying this is an open question: do such layers of abstraction that allow mechanism in the true sense exist? Complexity seems to be everywhere in biology, and my reaction so far has been to just throw up my hands up and say “it’s complicated!”. But (and this is another lesson learned from Rob), that’s not an excuse—we have to at least try. And I do think we can find some mechanistic wormholes through the seemingly infinite space of empiricism that we are currently mired in.
Regardless of what layers of abstraction we choose, however, I think that it is clear that a common feature of these future models will be that they are multifactorial, meaning that they will simultaneously incorporate the interactions of multiple molecules or cells or whatever the units we choose are. How do we deal with multiple interactions? I’m not alone in thinking that our models need to be quantitative, which as noted in my first post, is an idea that’s been around for some time now. However, I think that a fair charge is that in the early days of this field, our quantitative models were pretty much window dressing. I think (again a point that I’ve finally absorbed from Rob) that we have to start setting (and reporting) quantitative goals. We can’t pick and choose how our science is quantitative. If we have some pretty model for something, we better do the hard work to get the parameters we need, make hard quantitative predictions, and then stick to them. And if we don’t quantitatively get what we predict, we have to admit we were wrong. Not partly right, which is what we do now. Here’s the current playbook for a SysBio paper: quantitatively measure some phenomenon, make a nice model, predict that removal of factor X should send factor Y up by 4x, measure that it went up 2x, and put a bow on it and call it a day. I think we just have to admit that this is not good enough. This “pick and choose” mix of quantitative and qualitative analyses is hugely damaging because it makes it impossible to build upon these models. The problem is that qualitative reporting in, say, abstracts leads to people seeing “X affects Y” and “Y affects Z” and concluding “thus, X affects Z” even though the effects for X on Y and Y on Z may be small enough to make this conclusion pretty tenuous.
So I have a couple proposals. One is that in abstracts, every statement should include some sort of measure of the percentage of effect explained by the putative mechanism. I.e., you can’t just say “X affects Y”. You have to say something like “X explains 40% of the change in Y”. I know, this is hard to do, and requires thought about exactly what “explains” means. But yeah, science is hard work. Until we are honest about this, we’re always going to be “quantitative” biologists instead of true quantitative biologists.
Also, as a related grand challenge, I think it would be cool to try and be able to explain some regulatory process in biology out to 99.9%. As in, okay, we really now understand in some pretty solid way how something works. Like, we actually have mechanism in the true sense. You can argue that this number is arbitrary, and it is, but I think it could function well as an aspirational goal.
Any discussion of empiricism vs. theory will touch on the question of science vs. engineering. I would argue that—because we’re in an age of empiricism—most of what we’re doing in biology right now is probably best called engineering. Trying to make cells divide faster or turn into this cell or kill that other cell. And it’s true that look, whatever, if I can fix your heart, who cares if I have a theory of heart? One of my favorite stories along these lines is the story of how fracking was discovered, which was purely by accident (see Planet Money podcast): a desperate gas engineer looking to cut costs just kept cutting out an expensive chemical and seeing better yield until he just went with pure water and, voila, more gas than ever. Why? Who cares! Then again, think about how many mechanistic models went into, e.g., the design of the drills, transportation, everything else that goes into delivering energy. I think this highlights the fact that just like science and engineering are intertwined, so are mechanism and empiricism. Perhaps it’s time, though, to reconsider what we mean by mechanism to make it both more expansive and rigorous.
Personally, I think that these difficulties arise partially because of our tools, but mostly because I think we are still focused on the wrong layers of abstraction. Probably the most common current layers of abstraction are those of genes/molecules, cells, and organisms. Our most powerful models/mechanisms to date are the ones where we could draw straight lines connecting these up. Like, mutate this gene, make these cells look funny, now this person has this disease. However, I think these straight lines are more the exception than the norm. Mostly, I think these mappings are highly convoluted in interwoven systems, making it very hard to make predictions based on empiricism alone (future blog post coming on Omnigenic Model to discuss this further).
Which leads me to a proposal: let’s start thinking about other layers of abstraction. I think that the successes of the genes/molecules -> cells paradigm has led to a certain ossification of thought centered around thinking of genes and molecules and cells as being the right layers of abstraction. But maybe genes and cells are not such fundamental units as we think they are. In the context of multicellular organisms, perhaps cells themselves are passive players, and rather it is communities of cells that are the fundamental unit. Organoids could be a good example of this, dunno. Also, it is becoming clear that genetics has some pretty serious limits in terms of determining mechanism in the sense I’ve defined. Is there some other layer involving perhaps groups of genes? Sorry, not a particularly inspired idea, but whatever, something like that maybe. Part of thinking this way also means that we have to reconsider how we evaluate science. As Rob pointed out, we have gotten so used to equating “mechanism” to “molecules and their effects on cells” that we have become both closed minded to other potential types of mechanism while also deceiving ourselves into allowing empiricism to pose as mechanism under the guise of statistics. We just have to be open to new abstractions and not hold everyone to the "What's the molecule?" standard.
Of course, underlying this is an open question: do such layers of abstraction that allow mechanism in the true sense exist? Complexity seems to be everywhere in biology, and my reaction so far has been to just throw up my hands up and say “it’s complicated!”. But (and this is another lesson learned from Rob), that’s not an excuse—we have to at least try. And I do think we can find some mechanistic wormholes through the seemingly infinite space of empiricism that we are currently mired in.
Regardless of what layers of abstraction we choose, however, I think that it is clear that a common feature of these future models will be that they are multifactorial, meaning that they will simultaneously incorporate the interactions of multiple molecules or cells or whatever the units we choose are. How do we deal with multiple interactions? I’m not alone in thinking that our models need to be quantitative, which as noted in my first post, is an idea that’s been around for some time now. However, I think that a fair charge is that in the early days of this field, our quantitative models were pretty much window dressing. I think (again a point that I’ve finally absorbed from Rob) that we have to start setting (and reporting) quantitative goals. We can’t pick and choose how our science is quantitative. If we have some pretty model for something, we better do the hard work to get the parameters we need, make hard quantitative predictions, and then stick to them. And if we don’t quantitatively get what we predict, we have to admit we were wrong. Not partly right, which is what we do now. Here’s the current playbook for a SysBio paper: quantitatively measure some phenomenon, make a nice model, predict that removal of factor X should send factor Y up by 4x, measure that it went up 2x, and put a bow on it and call it a day. I think we just have to admit that this is not good enough. This “pick and choose” mix of quantitative and qualitative analyses is hugely damaging because it makes it impossible to build upon these models. The problem is that qualitative reporting in, say, abstracts leads to people seeing “X affects Y” and “Y affects Z” and concluding “thus, X affects Z” even though the effects for X on Y and Y on Z may be small enough to make this conclusion pretty tenuous.
So I have a couple proposals. One is that in abstracts, every statement should include some sort of measure of the percentage of effect explained by the putative mechanism. I.e., you can’t just say “X affects Y”. You have to say something like “X explains 40% of the change in Y”. I know, this is hard to do, and requires thought about exactly what “explains” means. But yeah, science is hard work. Until we are honest about this, we’re always going to be “quantitative” biologists instead of true quantitative biologists.
Also, as a related grand challenge, I think it would be cool to try and be able to explain some regulatory process in biology out to 99.9%. As in, okay, we really now understand in some pretty solid way how something works. Like, we actually have mechanism in the true sense. You can argue that this number is arbitrary, and it is, but I think it could function well as an aspirational goal.
Any discussion of empiricism vs. theory will touch on the question of science vs. engineering. I would argue that—because we’re in an age of empiricism—most of what we’re doing in biology right now is probably best called engineering. Trying to make cells divide faster or turn into this cell or kill that other cell. And it’s true that look, whatever, if I can fix your heart, who cares if I have a theory of heart? One of my favorite stories along these lines is the story of how fracking was discovered, which was purely by accident (see Planet Money podcast): a desperate gas engineer looking to cut costs just kept cutting out an expensive chemical and seeing better yield until he just went with pure water and, voila, more gas than ever. Why? Who cares! Then again, think about how many mechanistic models went into, e.g., the design of the drills, transportation, everything else that goes into delivering energy. I think this highlights the fact that just like science and engineering are intertwined, so are mechanism and empiricism. Perhaps it’s time, though, to reconsider what we mean by mechanism to make it both more expansive and rigorous.








